
How loss of TAFAZZIN function ignites mitochondrial dysfunction
Article title: Anomalous peroxidase activity of cytochrome c is the primary pathogenic target in Barth syndrome
Authors: Valerian E. Kagan, Yulia Y. Tyurina, Karolina Mikulska-Ruminska, Deena Damschroder, Eduardo Vieira Neto, Alessia Lasorsa, Alexander A. Kapralov, Vladimir A. Tyurin, Andrew A. Amoscato, Svetlana N. Samovich, Austin B. Souryavong, Haider H. Dar, Abu Ramim, Zhuqing Liang, Pablo Lazcano, Jiajia Ji, Michael W. Schmidtke, Kirill Kiselyov, Aybike Korkmaz, Georgy K. Vladimirov, Margarita A. Artyukhova, Pushpa Rampratap, Laura K. Cole, Ammanamanchi Niyatie, Emma-Kate Baker, Jim Peterson, Grant M. Hatch, Jeffrey Atkinson, Jerry Vockley, Bernhard Kühn, Robert Wessells, Patrick C. A. van der Wel, Ivet Bahar, Hülya Bayir & Miriam L. Greenberg
Journal: Nature Metabolism (Link to article)
***This paper is behind a paywall; please email Lindsay.marjoram@barthsyndrome.org if you would like a PDF of the article***
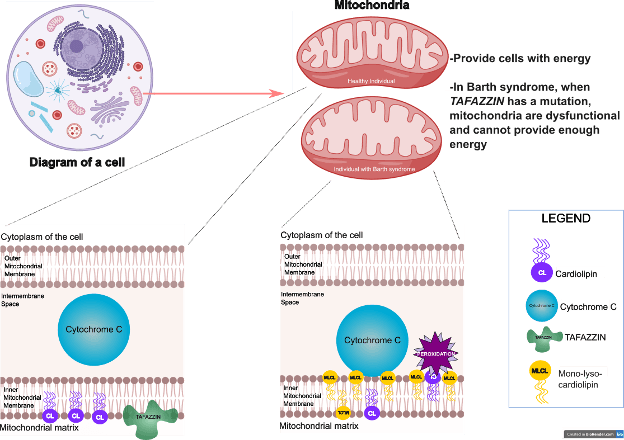
Barth syndrome, which is caused by changes or mutations in the gene TAFAZZIN (What is a mutation?), is a disease that affects the heart, skeletal muscle, and immune system. TAFAZZIN is located on the X-chromosome, which means that most cases of Barth syndrome occur in males (How do X-linked genes work?). Understanding precisely how a mutation in TAFAZZIN leads to Barth syndrome is critical for developing new treatments and better understanding how the disease occurs and progresses. Miriam Greenberg, PhD (member of Barth Syndrome Foundation’s Scientific and Medical Advisory Board and Scientific Liaison to the Board of Directors), Valerian Kagan, PhD, Hülya Bayir, MD, and scientific colleagues from the US and abroad recently published a paper in Nature Metabolism where they have, for the first time, defined a direct mechanism to explain how loss of TAFAZZIN function leads to structural and functional defects in the mitochondria. Defective mitochondria make less energy for cells and exhibit other cellular defects that impact tissues like the heart and skeletal muscle that contain large numbers of mitochondria.
Findings: In cells, TAFAZZIN is an important enzyme that helps make sure the correct version of a specialized fat called cardiolipin is incorporated into the membranes in the mitochondria. Mitochondrial membranes play many roles including structural support for the mitochondria but also act as a form of scaffolding on which many important chemical reactions occur, including reactions that produce energy for cells. When TAFAZZIN is mutated, cardiolipin levels are reduced and a precursor version of it (mono-lyso-cardiolipin or MLCL) is increased. While scientists know that high levels of the precursor mono-lyso-cardiolipin disrupt the mitochondria, the exact steps to get from point A (loss of functional TAFAZZIN) to point B (Barth syndrome symptoms) are not well understood. In this comprehensive and compelling paper, the authors show that when the precursor mono-lyso-cardiolipin is overproduced, it accumulates in the side of the lipid membrane putting the precursor in close proximity to a protein called cytochrome c. The interaction between mono-lyso-cardiolipin and cytochrome c changes the shape and behavior of cytochrome c and allows it to transform into something called a peroxidase, which causes toxic phospholipids to form.
Peroxidation of the already scarce mature cardiolipin as well as the precursor mono-lyso-cardiolipin and other phospholipids damages the lipids in the mitochondria. The conversion of cytochrome c to a peroxidase after interaction with mono-lyso-cardiolipin was shown in experiments on yeast cells lacking Tafazzin, mouse cells with no or reduced Tafazzin, cells obtained from patients with Barth syndrome, heart tissue from Barth syndrome patients with mutated TAFAZZIN, and in a fruit fly model of Barth syndrome (Why the fly?). In cell experiments and experiments with fruit flies, the research team showed that administration of a compound called imidazole oleic acid blocks the peroxidase activity of cytochrome c and in fruit flies, prevented the fatigue caused by loss of TAFAZZIN.
Take-Home Message: For the first time, researchers have demonstrated that high levels of the cardiolipin precursor, mono-lyso-cardiolipin, that result from loss of TAFAZZIN lead to dysfunctional organization of the mitochondrial lipid membrane and drive an abnormal interaction of mono-lyso-cardiolipin with cytochrome c. This interaction causes cytochrome c to generate toxic lipid products that lead to reduced energy production and other mitochondrial dysfunctions. Imidazole oleic acid, a compound that blocks the toxic activities of cytochrome c, can lead to improvements in fatigue in a fruit fly model of Barth syndrome. In future studies, the authors will try to identify the most effective inhibitors of the peroxidation driven by cytochrome c and test them in a mouse model of Barth syndrome. These future studies have the potential to generate a new class of therapeutics for the treatment of Barth syndrome.


